The “Living Drug” For Cancer
Apr 11th 2022
The rising burden of cancer worldwide has led to active research in order to understand the molecular biology of cancer progression and to find new cures for this grave disease. Traditionally, the major approaches to cancer treatment was surgery, radiotherapy, chemotherapy, small molecule targeted drugs, monoclonal antibodies and hematopoietic stem cell transplantation. Surgical removal can benefit only early-stage patients whereas radiotherapy and chemotherapy have very poor selectivity and can damage normal tissues. Small molecules and monoclonal antibodies have shown better efficacy and lower toxicity but have resulted in gene mutation of tumor cells and drug tolerance in many patients. Finding a donor makes hematopoietic stem cell transplantation often a difficult option. Cellular immunotherapy is the latest approach that has shown excellent clinical outcomes and has been the forefront of cancer therapy in recent times.
1. Types of immunotherapeutic approaches
It is not very long back when scientist discovered that cancer cells can evade the body’s immune surveillance. Immunotherapy has been the additional line of treatment in which the patient’s own immune system in boosted to recognize and destroy cancer cells. Over the years scientists have investigated various strategies in which this can be achieved.
1.1 Non-specific immunotherapies
This type of treatment uses proteins of the innate immune system to heighten body’s immunity. Interferons are cytokines that are being used to slow down the growth of cancer cells. Interferon alpha (Roferon-A [2a], Intron A [2b], Alferon [2a]) are the commercially produced interferons that come under cancer drugs. Interleukin-2, is another type of cytokine that has proven to stimulate T-cell production which is effective in cancer treatment. IL-2 under the brand name Proleukin (Aldesleukin) is mainly used to treat kidney cancer and skin cancer, including melanoma.
1.2 Immune checkpoint inhibitors
Recent developments in antibodies as immune checkpoint inhibitors have demonstrated good success rates in clinical trials. These antibodies work by blocking the molecular pathways involved in suppressing immune functions. Many cancers have shown to use immune checkpoint pathways like PD-1/PDL-1 and CTLA-4 to escape the immune system. Drugs like Ipilimumab (anti-CTLA4) and Nivolumab (anti-PD1) are mAbs that block these pathways enhancing T cell response. There is a lot of active research undergoing in this area to investigate various immune-suppression pathways used by cancer cells. In addition, a newer class of mAbs known as bispecific T-cell enhancing antibodies has been designed to simultaneously target a tumor-associated antigen (TAA) and a T-cell antigen (e.g., CD19 and CD3,respectively), which results in enhanced co-localization of T cells and tumors (Dahlen E et al., 2018).
1.3 Oncolytic virus therapy
This method uses genetically modified viruses that can specifically target tumor cells as host. The viral life cycle results in the lysis of the cancer cells and release of tumor-specific antigens inducing an immune response. T-Vec (talimogene laherparepvec), a second-generation oncolytic herpes simplex virus type 1 (HSV-1) armed with GM-CSF, was recently approved as the first oncolytic virus drug for melanoma in the USA and Europe. (Fukuhara et al., 2016). Other oncolytic viruses under development are vaccinia virus JX-594 (pexastimogene devacirepvec) for hepatocellular carcinoma, GM-CSF expressing adenovirus CG0070 for bladder cancer, and Reolysin (pelareorep), a wild-type variant of reovirus, for head and neck cancer. One of the major challenges with using viruses is the presence of circulating anti-viral antibodies that can neutralise the virus before they reach the tumor site. Hence, intralesional administration is the preferred over an intravenous injection. Oncolytic viral therapy show promising results in clinical trials and could soon be a popular scheme for treating cancers.
1.4 Cancer vaccines
Preventive and treatment cancer vaccines have been developed that can elicit an immune response in patients. Preventive vaccines include viral vaccines like HPV or Hepatitis B vaccine that can prevent infection of cancer-causing viruses. Therapeutic cancer vaccines, on the other hand, are used for treatment of cancer. Two types of therapeutic vaccines have been developed:
- Autologous vaccines
It is a personalized vaccine made from an individual’s own cells which can be a cancer or immune cell. In the first approach, cancer cells are isolated from the patient, irradiated and re-introduced into the body along with BCG immune adjuvant to stimulate an immune response. This has shown prolonged expression and release of tumor associated antigens (TAA) to allow sufficient uptake by and activation of immune effector cells (de Gruijl et al., 2008).
Another approach is to use an individual’s own immune cells to make the vaccine. The US FDA has licensed Sipuleucel-t (Provenge®), an autologous vaccine made from the patient’s dendritic cells. It has shown in clinical trials to extend overall survival in treatment-resistant metastatic prostate cancer cases.
- Allogenic vaccines
These are vaccines generated from “non-self” or human cancer cell lines to induce antitumor immunity in patients through TAA presentation. Even though, allogeneic vaccines are cost-effective and easy to develop, clinical trials have not shown much efficacy owing to the tumor heterogeneity found among cancer patients.
Apart from whole-cell cancer vaccines mentioned above, protein, peptide and DNA vaccines are also being extensively studied for cancer therapeutic interventions.
1.5 Adoptive T-cell therapy
This type of immunotherapy involves ex-vivo expansion of the patient’s own T-cells and then infusing it back into the patient. These T-cells can be taken from the tumor region (known as tumor infiltrating lymphocytes or TILs that are capable of recognizing tumor antigens) or can be normal T-cells which are re-engineered in-vitro to target cancer cells. The latter is called chimeric antigen receptor (CAR) T-cell therapy as the cells are genetically engineered to produce a membrane spanning fusion receptors with defined specificities for TAAs. CAR T therapy has been very effective in the treatment of lymphoid malignancies. Many research groups are actively working on ways of modifying T cells to treat solid tumors as well.
Immunotherapy is a rapidly developing field and of late, doctors and scientists are looking at combination therapy approaches to increase the efficacy of most treatments. For example, oncolytic virus along with immune checkpoint inhibitors demonstrated better response and survival in most patients. Likewise, a combination of two or more immune-checkpoint inhibitors and vaccine can considerably slow down disease progression.
In fact, immunotherapy is a personalized treatment approach and needs to be tailor-made for each patient. Moreover, all patients might not benefit from immunotherapy. Rigorous diagnostic tests need to be run that can detect which kind of therapy will be the most effective in every case.
2. CAR T-cell therapy
Adoptive T-cell therapy or Adoptive cell transfer (ACT) is often referred to as a “living drug” since, the actual drug is a living T-cell. ACTs include the use of TILs, TCRs or CARs and by far CAR has been the most advanced. Chimeric Antigen Receptor (CAR) T-cell therapy has become one of the most powerful strategy for blood cancers in patients who don’t respond to other therapies. These re-programmed T-cells are capable of eliciting a very specific and robust anti-tumor response. In 2017, US FDA approved the first immunotherapy for acute lymphoblastic leukemia (ALL) using CD19-targeted CAR T-cells.Tisagenlecleucel (KYMRIAH, Novartis) is another FDA-approved CAR T cell therapy for pediatric patients with recurrent B cell acute lymphoblastic leukemia (ALL) after. Axicabtagene ciloleucel (YESCARTA, Kite Pharmaceuticals) has also been approved to treat relapsed/refractory diffuse large B-cell lymphoma (DLBCL) in the year 2017. Several more CARs are in the pipeline for FDA approvals. This success has prompted CAR T cell approaches for diseases other than cancer including regulatory CAR T cells for autoimmune disease and tissue transplantation. Likewise, there are multiple research groups working on allogeneic CAR T cells treatment approaches.
2.1 How is it done?
The production of CAR-T cells requires isolation and re-engineering of T cells from the patient’s blood. To begin with, leukocytes are separated from blood, a process known as leukocyte apheresis. This is one of the most critical steps as the leukapheresis products from cancer patients consist of a heterogeneous population of cells, including T cells, myeloid cells (e.g., monocytes, neutrophils, and dendritic cells), natural killer cells, erythroid cells, and malignant cells. This is followed by elutriation to remove myeloid cells, T lymphocyte enrichment and transgene delivery.
Usually, the peripheral blood mononuclear cells (PBMCs) are isolated and transferred to a cell-processing center. Here, the specific T cells are selected and stimulated to actively proliferate using the cytokine interleukin 2 (IL-2) and anti-CD3 antibodies. The purified T-cells are then transduced with a gene encoding the engineered CAR via a retroviral vector, the CRISPR/Cas9 tool or of late, the Sleeping Beauty transposon system.
Meanwhile, the patient undergoes lymphodepletion chemotherapy prior to the introduction of the engineered CAR-T cells. The reduction of circulating leukocytes in the blood will upregulate cytokine production thus reducing the competition for resources. This environment ultimately helps to promote the expansion of the engineered CAR-T cells.
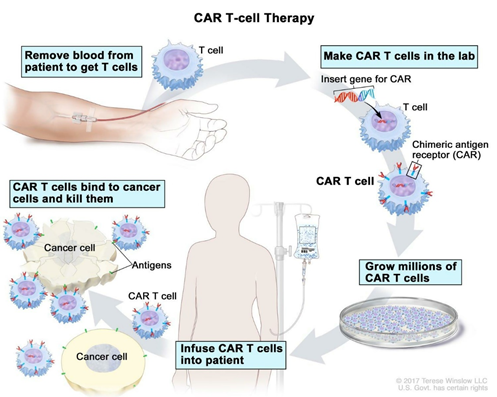
Figure 1: CAR T cell therapy procedure. Reference: www.cancer.gov
The reengineered CAR T cells are expanded in the lab and frozen and sent to the hospital or center where the patient is being treated. These cells are thawed and then infused into the patient’s bloodstream where they multiply in number and also create memory cells. These cells specifically recognize and attack cells that have the targeted antigen on their surface. CAR T cells may eradicate all of the cancer cells and may remain in the body months after the infusion has been completed. The therapy has resulted in long-term remissions for some types of blood cancer. Moreover, it is important to attain efficient T-cell purification and tumor cell purging to improve the safety and potency of cellular products for adoptive transfer therapies.
2.2 Chimeric Antigen Receptors (CARs)
Rs are genetically engineered to express an extracellular domain for tumor antigen recognition, linked to one or more intracellular signalling domains that mediate T-cell activation. CARs include three parts: an extracellular antigen recognition domain of the single-chain Fragment variant (scFv) derived from an antibody), a transmembrane domain and an intracellular T cell activation domain of CD3ζ (Zhang et al., 2017). The scFv is the antigen recognition molecule that consists of the variable heavy (VH) and variable light (VL) chains of an antibody, fused by a peptide spacer of ~15 residues in length. This molecule is joined to an intracellular signalling molecule comprised of the TCR T3 zeta chain (CD3ζ) subunit, which is an activation-signalling domain. The endodomain can also be another immune- receptor-tyrosine-based-activation-motif [ITAM]-containing protein), and an optional tandem co-stimulation domains such as the 4-1BB or CD28 signalling modules (Sermer et al., 2019).
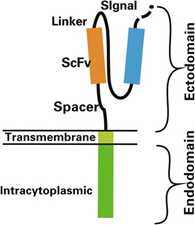
Figure 2: Structure of chimeric antigen receptor (CAR). Reference: Zhang et al., 2017
The CAR T cells have been divided into four generations since their initial development in 1989. Preclinical studies using first-generation CAR T cells directed against CD19 expressing B-cell malignancies and HER2/Neu showed promising results. The second-generation CAR T cells were developed by incorporating an additional costimulatory signalling domains such as CD28 or 4–1BB in the intracellular domain. This provided an amplified T-cell activation signal which further improved the efficacy of CAR T cells.
Further optimization led to the development of the third-generation CAR T cells, which incorporated two-tandem costimulatory domains by coupling ICOS, CD27, CD28, 4–1BB, or OX40 (e.g., CD28/4–1BB/CD3ζ or CD28/OX40/CD3ζ), hence demonstrating varying degrees of in vitro and in vivo T-cell activation, proliferation, and cytokine (interleukin [IL]-2) production (Sharma et al., 2019). The recently developed fourth-generation of armored CAR T cell incorporates not only the two-costimulatory domains but also an additional transgene for cytokines (e.g., IL-12) or ligands (e.g., CD40L or 4–1BBL). This equips these CAR T cells to survive and/or disrupt an immunosuppressive tumor microenvironment.
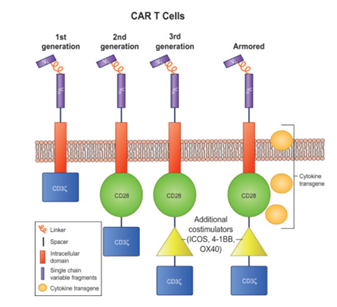
Figure 3: Different generations of CAR T cell constructs.
The 1st generation CAR T comprising of a T cell signalling domain; the 2nd generation, with one costimulation domain and a T cell signalling domain; the 3rd generation, with two costimulation domains and a T cell signalling domain; and the armored CAR T cell with similar construct to third generation but with an additional pro-inflammatory cytokine transgene.
Reference: Sharma et al., 2019
2.3 Side Effects Of CAR T cell Therapy
Most patients on CAR T cell therapy suffer from severe toxicities mainly, cytokine release syndrome (CRS) and neurotoxicity (or CAR T cell‐related encephalopathy syndrome [CRES]). It has been observed that cells with CD28 co-stimulatory domain show a greater incidence of neurotoxicity (NT) than CRS, whereas the opposite is seen in cells with 4-1BB.
CRS mimics a systemic inflammatory response syndrome (SIRS), which may manifest with fever, hemodynamic instability, hypoxia, and end‐organ dysfunction. The degree of severity may correlate with CAR T‐cell infusion dose, extent of expansion of cells in vivo, and amount of tumor burden (Sermer et al., 2019). IL‐6 receptor blockers such as tocilizumab and siltuximab was approved by the US FDA for treatment of CRS occurring after CAR T‐cell therapy. Notably, neither of these blockers affected the efficacy of the therapy.
The occurrence of CNS toxicity ranges from 0 to 87% and seems to be most frequent in immature B cell diseases. The clinical features range from headache, pain, memory loss, meningismus, dizziness, alterations in mental status, movement disorders, impaired speech, seizures and encephalopathy to coma (Yanez L et al., 2019). Patients who experience neurotoxicity are monitored closely. The management of CNS toxicity is usually done using corticosteroids. It has been reported that neurotoxicity does not respond to anti IL-6 blockade in most patients and tocilizumab usually worsens the condition. Moreover, neurotoxicity takes longer time to resolve when compared to CRS.
Besides, side effects like tumor lysis syndrome, cytopenias, hypogammaglobulinemia, anemia, and cardiac toxicity are also seen in many patients. For these reasons, administration of CAR T cell therapies requires specialized training under the FDA Risk Evaluation and Mitigation Strategies to manage adverse events. Active scientific research is underway in order to create better CAR designs that can have less toxicity and better potency.
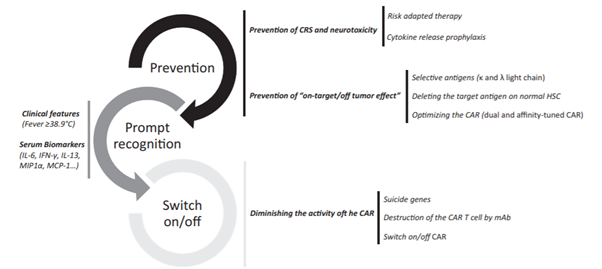
Figure 4: Future strategies to decrease side effects related with CAR T cell therapy. Reference: Yáñez et al., 2019
3. Treating Solid Tumors
CAR-redirected T cell therapies have been very effective in hematologic malignancies but the success rate has been low in treating patients with solid tumors. For solid tumors, immunotherapy based on TIL generation or immune cell blockade has shown better clinical efficacy.
Most of the solid tumors arise from epithelial origin and express antigen heterogeneity, making selection of a specific TAA very difficult. As a result most of the CARs designed for solid tumors resulted in severe off-tumor toxicities leading to organ failures. Even CARs for antigens identified as tumor specific has presented poor efficacy in clinic in spite of their lower toxicities. This could be attributed to poor trafficking to the tumor site, limited persistence and proliferation within the host or immunosuppression within the hostile tumor microenvironment.
However, in a recent study, an end-stage patient with recurrent glioblastoma showed positive response when treated with CAR T cells engineered against IL-13Rα. In another trial, anti-disialoganglioside GD2 CAR T-cells have been used to treat evaluable paediatric patients with neuroblastoma, where 3 of 11 patients with active disease achieved complete remission. These outcomes have emphasized the potential of CAR T cell therapies and the need for further development.
According
to the three-pronged approach, proposed by Knochelmann et al., a multi-faceted
attack on solid tumors resistant to standard CAR T cell therapies may
improve their efficacy in clinical trials. This can be achieved by ensuring
that the therapeutic strategy should encompass three axes: (1) a CAR with high
fidelity targeting of more than one tumor antigen and trafficking capacity, (2)
selection of a T cell subset with potent self-renewal and migratory
capacity for long-term persistence and immunity, and (3) ability to harness and
rejuvenate the host response to tumor neoantigens. Till date, a single arm
(CAR, subset, or host response) has not been sufficient for long-term responses
against aggressive solid tumors in clinical studies (Knochelmann et al., 2018).
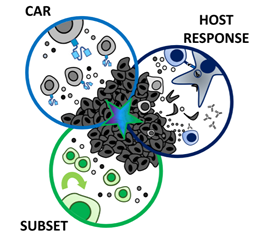
Figure 5: Three-pronged approach to improve chimeric antigen receptor (CAR) T cell therapies in solid tumors. Reference: Knochelmann et al., 2019
Several new CAR designs are being currently tested to improve efficacy in solid tumors. Dual CARs for multiple antigen recognition is expected to improve tumor infiltration, whereas, BBζ CAR T cells with increased mitochondria can overcome hypoxic TME. Another interesting approach based on the clinical success of CAR and immune blockade combination therapy is the design of CAR T cells that have been engineered to secrete checkpoint inhibitor antibodies themselves. Anti CAIX CAR T cells engineered to secrete anti-PD-L1 antibodies and CAR19 T cell designed to constitutively secrete anti-PD-1 have shown significant anti-tumor activity in mouse models (Martinez and Moon, 2019). CAR T cells with inducible suicide genes are also being tested as a strategy to improve safety of CAR T cell therapy.
The Road Ahead
Immunotherapy has truly revolutionised the way cancer can be treated. After the success of immune checkpoint blockade therapy, adoptive T cell therapies have emerged as the next big thing.
CAR T cell therapy has been fruitful in haematological malignancies, however it is still naïve and further development is required to improve potency in solid tumors and to make it commercially viable. Significant research is ongoing in terms of identifying target antigens, avoiding toxicity, improving CAR T cell trafficking and entry into the tumor site, and promoting better signalling, less exhaustion, and memory phenotypes in solid tumors. There is also an urgent need for identification of biomarkers to enable clinicians to determine which patients will respond to CAR T-cell treatment so that it can be tweaked for maximization of therapeutic effects and better toxicity management.
With the advent of tools like CRISPR/Cas9 and TALENS, scientists are also exploring the options of creating allogeneic CAR T cell, possibly derived from stem cells that could one day become an ‘off-the shelf’ T cell drug.
In this progressive era of personalised medicine, CAR technology is undoubtedly a medical breakthrough. A greater understanding of host immune system and tumor biology will help enhance the standards of cellular therapies. Conclusively, CAR T cell therapies has opened up promising avenues for therapeutic strategies not only in oncology but also in other fields including regenerative medicine.
References
Andy J. Minn. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015 November ; 36(11): 725–737. doi:10.1016/j.it.2015.09.007.
CE Brown and PS Adusumilli. Next frontiers in CAR T-cell therapy. Molecular Therapy — Oncolytics (2016) 3, 16028; doi:10.1038/mto.2016.28
Cheng Zhang, Jun Liu, Jiang F. Zhong, and Xi Zhang. Engineering CAR-T cells. Biomark Res. 2017; 5: 22. doi: 10.1186/s40364-017-0102-y
Dahlen E, Veitonmaki N, Norlen P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. 2018;6(1):3-17.
Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019 May;94(S1):S3-S9. doi: 10.1002/ajh.25418. Epub 2019 Feb 18.
Hiroshi Fukuhara, Yasushi Ino and Tomoki Todo. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci 107 (2016) 1373–1379. doi: 10.1111/cas.13027
Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S and Paulos CM (2018) CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 9:1740. doi: 10.3389/fimmu.2018.01740
Kosti P, Maher J and Arnold JN (2018) Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 9:1104. doi: 10.3389/fimmu.2018.01104
Martinez M and Moon EK (2019) CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 10:128. doi: 10.3389/fimmu.2019.00128.
Next frontiers in CAR T-cell therapy . Molecular Therapy — Oncolytics (2016) 3, 16028; doi:10.1038/mto.2016.28
Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017 May 24;545(7655):423-431. doi: 10.1038/nature22395.
Sanjay Srivatsan, Jaina M Patel, Erica N Bozeman, Imade E Imasuen, Sara He, Danielle Daniels, and Periasamy Selvaraj. Allogeneic tumor cell vaccines. Hum Vaccin Immunother. 2014 Jan 1; 10(1): 52–63. doi: 10.4161/hv.26568.
Sermer D, Brentjens R. CAR T-cell therapy: Full speed ahead. Hematol Oncol. 2019 Jun;37 Suppl 1:95-100. doi: 10.1002/hon.2591.
Steven Feins, Weimin Kong, Erik F. Williams, Michael C. Milone, Joseph A. Fraietta. An introduction to chimeric antigen receptor (CAR) T-cellimmunotherapy for human cancer. Am J Hematol. 2019 DOI: 10.1002/ajh.25418.
Tanja D. de Gruijl, Alfons J. M. van den Eertwegh, Herbert M. Pinedo, Rik J. Scheper. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother (2008) 57:1569–1577 DOI 10.1007/s00262-008-0536-z.
Xiuyan Wang, Isabelle Rivière. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Molecular Therapy Oncolytics. Volume 3, 2016, 16015. doi.org/10.1038/mto.2016.15.
Yanez L, Sanchez-Escamilla M, Perales MA. CAR T Cell Toxicity: Current Management and Future Directions. HemaSphere, 2019;3:2. http://dx. doi.org/10.1097/HS9.0000000000000186
Written By: Shalitha Sasi